Ver traducción automática
Esta es una traducción automática. Para ver el texto original en inglés haga clic aquí
#Tendencias de productos
{{{sourceTextContent.title}}}
¿Cuándo entró en vigor la Parte 11 del Título 21 del CFR?: en qué consiste y cómo cumplir con sus requisitos
{{{sourceTextContent.subTitle}}}
El cumplimiento de la Parte 11 del Título 21 del Código de Regulaciones Federales (21 CFR) es obligatorio para el sector farmacéutico y de los complementos alimenticios regulado por la FDA. Si no sabes en qué consiste ni cómo cumplir con esta normativa, encontrarás las respuestas en este texto.
{{{sourceTextContent.description}}}
En los sectores industriales regulados, los registros digitales ya no son simples archivos utilizados para almacenar datos. Se han convertido en fuentes de pruebas trazables.
Para las empresas farmacéuticas, nutracéuticas, de dispositivos médicos y de fabricación por contrato, los registros electrónicos y las firmas electrónicas son esenciales para garantizar la seguridad de la producción, mantener el control de calidad y prevenir costosas interrupciones causadas por cambios en la formulación o ajustes en los parámetros del proceso. A medida que la normativa estadounidense ha ido evolucionando, el cumplimiento de la norma 21 CFR Parte 11 se ha convertido gradualmente en un requisito obligatorio, en lugar de una inversión opcional.
Para muchos fabricantes, especialmente las pequeñas y medianas empresas, la implementación de soluciones de validación de sistemas y de gestión de datos conformes puede parecer costosa y compleja. Como resultado, a menudo surgen primero dos preguntas clave: ¿cuándo entró en vigor la norma 21 CFR Parte 11 y en qué consiste el cumplimiento de la norma 21 CFR Parte 11?
Esta guía exhaustiva ofrece una explicación detallada de la norma 21 CFR Parte 11. Analizaremos qué es la norma 21 CFR Parte 11, quién debe cumplirla, cuáles son sus requisitos clave y cómo lograr el cumplimiento de la norma 21 CFR Parte 11 de una manera práctica y rentable.
Puntos clave
Cuándo entró en vigor la Parte 11 del Título 21 del CFR y su contexto histórico.
Qué es la Parte 11 del Título 21 del CFR en la industria farmacéutica.
Quién debe cumplir con la Parte 11 del Título 21 del CFR en diversos sectores.
Requisitos fundamentales de la Parte 11 del 21 CFR en materia de registros, firmas, acceso y registros de auditoría.
Cómo cumplir con la Parte 11 del 21 CFR paso a paso.
Cómo los equipos que cumplen con la Parte 11 respaldan la producción farmacéutica y nutracéutica.
1. ¿Cuándo entró en vigor la norma 21 CFR Parte 11?
Según la página web oficial de la FDA, la norma 21 CFR Parte 11 entró en vigor el 20 de agosto de 1997.
¿Qué motivó la promulgación de esta normativa? Sus orígenes se remontan a la gran transición de los registros en papel a los sistemas informatizados. Anteriormente, los registros de producción farmacéutica, los informes de laboratorio y los documentos de calidad se creaban, firmaban y almacenaban en su mayor parte en papel. A medida que la producción se fue ampliando, los simples registros en papel dejaron de satisfacer la necesidad de una documentación rápida y una recuperación inmediata de los datos, lo que llevó a las empresas a adoptar sistemas digitales.
Para garantizar la fiabilidad de estos nuevos sistemas, la Parte 11 del Título 21 del Código de Regulaciones Federales (21 CFR) somete los registros electrónicos y las firmas electrónicas a una estricta supervisión normativa. Cuando las empresas utilizan datos digitales para respaldar decisiones sujetas a la normativa de la FDA, dichos datos deben ser precisos y seguros, además de trazables y recuperables durante las inspecciones de la FDA.
2. ¿Qué es el 21 CFR, parte 11?
Definición del 21 CFR, parte 11
En esencia, ¿qué es el 21 CFR, parte 11? Se trata de una normativa establecida por la FDA de EE. UU. que dicta cómo gestionar los registros electrónicos y las firmas.
Para comprender la normativa 21 CFR Parte 11, pensemos en la supervisión histórica que ejercía la FDA sobre los archivadores cerrados con llave, controlando quién los gestionaba y haciendo un seguimiento de quién abría o modificaba su contenido. A medida que estos archivadores han evolucionado hacia sistemas digitales, la FDA ha introducido estas directrices de la 21 CFR Parte 11 para garantizar que los sistemas estén debidamente controlados y que los datos permanezcan seguros.
¿Qué tipos de registros se incluyen?
Los registros electrónicos sujetos a la norma 21 CFR Parte 11 pueden incluir:
Registros electrónicos de lotes
Registros de fabricación
Registros de control de calidad
Registros de funcionamiento de los equipos
Resultados de ensayos de laboratorio
Registros de limpieza y mantenimiento
Registros de formación
Registros de control de cambios y desviaciones
Cabe señalar que esta normativa no se aplica a todos los archivos digitales, sino únicamente cuando los registros electrónicos se utilizan para cumplir los requisitos de mantenimiento de registros de la FDA.
¿Qué sectores deben cumplirla?
Los requisitos de cumplimiento establecidos en el título 21 del CFR, parte 11, pueden aplicarse a una amplia gama de entidades, entre las que se incluyen los fabricantes de productos farmacéuticos, suplementos dietéticos y productos sanitarios, así como a las organizaciones de fabricación por contrato, los laboratorios de ensayo y las empresas que exportan a Estados Unidos productos regulados por la FDA.
Por consiguiente, preguntas como «¿Se aplica el Título 21 del CFR, Parte 11, a los productos sanitarios?» y «¿Qué abarca el Título 21 del CFR, Parte 11?» son fundamentales; el principio fundamental que rige la aplicabilidad de la norma es que los requisitos de cumplimiento se activan siempre que se utilicen sistemas electrónicos para gestionar registros o autorizaciones sujetos a los requisitos de integridad de datos de la FDA.
3. ¿Por qué se introdujo la Parte 11 del Título 21 del CFR?
La Parte 11 del Título 21 del CFR se introdujo porque la fabricación digital generaba tanto nuevas eficiencias como nuevos riesgos de cumplimiento.
La Parte 11 del Título 21 del CFR surgió como respuesta a las mejoras en la eficiencia y a los nuevos riesgos de cumplimiento asociados a la fabricación digital. A medida que la industria pasaba de los registros en papel a los sistemas digitales, la FDA necesitaba establecer una normativa que garantizara la fiabilidad de los registros electrónicos; esta norma se aplica a los registros regulados por la FDA que se crean, modifican, mantienen, archivan, recuperan o transmiten electrónicamente.
El objetivo principal de la norma es salvaguardar la integridad de los datos de la Parte 11 del Título 21 del CFR y evitar la manipulación o la falsificación, exigiendo que los sistemas cuenten con registros de auditoría, que actúan de forma muy similar a una «caja negra» que registra la secuencia real de los acontecimientos. Durante las inspecciones de la FDA, los reguladores examinan los registros electrónicos de lotes, los requisitos de la pista de auditoría de la Parte 11, los controles de acceso de los usuarios y las firmas electrónicas conformes con la Parte 11 del Título 21 del CFR; estos registros históricos exhaustivos constituyen la piedra angular del cumplimiento normativo, la calidad del producto y la confianza de los clientes.
4. ¿Quién debe cumplir con la Parte 11 del Título 21 del CFR?
No todas las empresas necesitan el mismo nivel de control, pero muchos fabricantes regulados por la FDA deberían evaluar quién necesita cumplir con la Parte 11 del Título 21 del CFR en función de sus sistemas y su mercado.
Fabricantes de medicamentos
En el sector farmacéutico, los sistemas de registros electrónicos que incluyen información sobre lotes, control de calidad, registros de equipos o autorizaciones de liberación suelen estar sujetos a la Parte 11 del 21 CFR. Sin embargo, una vez que los registros electrónicos sustituyen oficialmente a los registros en papel, esta obligación de cumplimiento pasa a ser obligatoria en lugar de opcional.
Fabricantes de complementos alimenticios
Si un fabricante de complementos alimenticios gestiona electrónicamente los registros de producción, calidad o lotes y el producto está destinado a la venta en Estados Unidos, es probable que se le aplique el cumplimiento de la Parte 11 del CFR.
Fabricantes de productos sanitarios
Para quienes se preguntan si la Parte 11 del Título 21 del CFR se aplica a los productos sanitarios, la respuesta es sí cuando los registros electrónicos respaldan los requisitos del sistema de calidad de la FDA.
Organizaciones de fabricación por contrato
Las organizaciones de fabricación por contrato (CMO) suelen necesitar un estricto cumplimiento de la Parte 11 del Título 21 del CFR de la FDA, ya que prestan servicio a múltiples clientes y se enfrentan a frecuentes auditorías de los mismos.
Empresas que exportan a Estados Unidos
Incluso si el fabricante está ubicado fuera de Estados Unidos, por lo general sigue siendo obligatorio cumplir con los requisitos de la Parte 11, siempre que los productos se vendan en EE. UU. y los registros electrónicos se refieran a actividades reguladas por la FDA.
5. ¿Cuáles son los requisitos de la Parte 11 del 21 CFR?
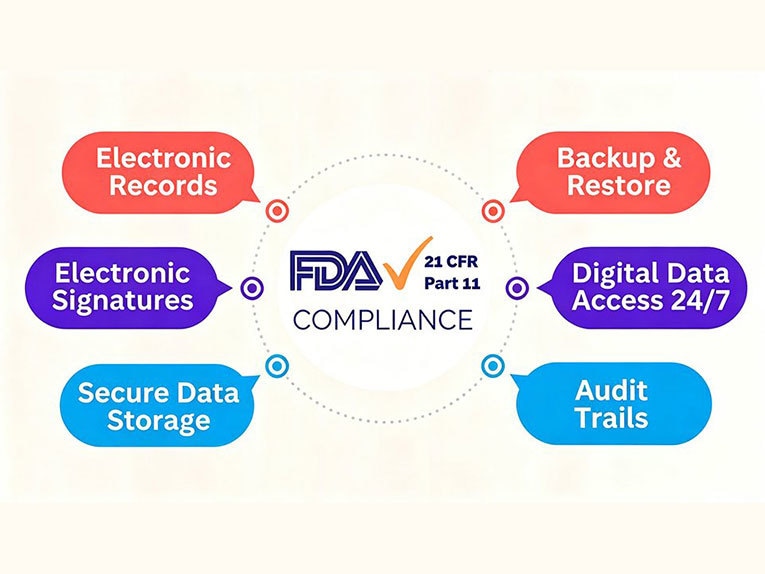
Pues bien, según el documento, ¿cuáles son exactamente los requisitos de la Parte 11 del 21 CFR? La respuesta puede agruparse en varios controles prácticos. Los requisitos de cumplimiento de la Parte 11 del 21 CFR se centran en cinco áreas clave: registros electrónicos, firmas electrónicas, pistas de auditoría, controles de acceso de los usuarios y conservación y recuperación de registros.
Registros electrónicos
Los requisitos de la Parte 11 del Título 21 del CFR para los registros electrónicos establecen que estos deben ser precisos, completos, seguros, legibles y recuperables. Las empresas deben garantizar una conservación adecuada de los registros y un almacenamiento seguro de los datos.
Firmas electrónicas
Las firmas electrónicas según la Parte 11 de la FDA deben establecer claramente la identidad y la responsabilidad, mostrando el nombre del firmante, la marca de tiempo y el significado de la acción (por ejemplo, «aprobación para su lanzamiento»), a fin de garantizar que tengan la misma validez legal que las firmas manuscritas.
Registros de auditoría
Una pregunta habitual es cómo funcionan los registros de auditoría en los equipos farmacéuticos. Los registros de auditoría registran automáticamente todas las acciones críticas, incluidas la creación, la modificación, la eliminación, la aprobación y los cambios en el sistema, para garantizar la trazabilidad. Si los datos de peso de un lote se modifican de 500 kg a 510 kg, el registro de auditoría recoge quién realizó la modificación, la hora en que se produjo y los valores antes y después de la modificación, lo que facilita el análisis de la causa raíz.
Controles de acceso de usuarios
Los controles de acceso garantizan que solo el personal autorizado pueda realizar acciones específicas mediante la protección con contraseña y la gestión de permisos, utilizando la autenticación multifactorial y el principio del privilegio mínimo.
Conservación y recuperación de registros
Los registros deben permanecer fácilmente accesibles durante todo el periodo de conservación exigido por la FDA; la imposibilidad de recuperar los datos con prontitud durante una auditoría se considera una grave infracción de cumplimiento.
6. Cómo cumplir con la norma 21 CFR Parte 11
Lograr el cumplimiento normativo es una de las principales preocupaciones para la mayoría de las empresas farmacéuticas y de productos sanitarios. En esta sección, analizaremos el tema en profundidad, abordando los pasos específicos de implementación, los posibles problemas y las soluciones correspondientes.
Paso 1: Realizar una evaluación de cumplimiento de la norma 21 CFR Parte 11
Debe comenzar comparando todos los sistemas que crean, modifican, almacenan, recuperan o transmiten registros electrónicos regulados por la FDA con las directrices de la norma 21 CFR Parte 11 para productos farmacéuticos. La evaluación debe incluir la validación, el registro de auditoría, el control de acceso de los usuarios, las firmas electrónicas, la conservación de registros y la seguridad.
Seguir la lista de comprobación de cumplimiento del 21 CFR Parte 11 puede agilizar el proceso.
Paso 2: validar los sistemas informatizados
Un sistema que cumpla con el 21 CFR Parte 11 debe someterse a un proceso de validación para demostrar que funciona según lo previsto. En la práctica, la validación debe llevarse a cabo como una secuencia estructurada de acciones:
1. Definir y evaluar el alcance y el riesgo del sistema
Empiece por documentar claramente cuál es la función del sistema e identificar dónde se encuentra el riesgo. Por ejemplo, una máquina de llenado de cápsulas debe registrar el peso de llenado, la velocidad y el número de lote.
2. Probar y verificar el rendimiento del sistema
A continuación, realice pruebas estructuradas mediante IQ, OQ y PQ. Esta evaluación sirve para comprobar si el sistema funciona según lo esperado.
3. Documentar, aprobar y mantener el control
Por último, todas las actividades de validación deben documentarse, revisarse y aprobarse antes de la puesta en servicio del sistema.
Paso 3: Implementar la funcionalidad de registro de auditoría
Debe garantizar la integridad de la función de registro de auditoría, confirmando que el sistema almacena y registra de forma segura los datos —como las marcas de tiempo y los cambios— y permite su recuperación precisa. Tomemos como ejemplo el ajuste de los parámetros de una máquina de llenado de cápsulas: una vez que un operador modifica los parámetros de llenado, el sistema captura automáticamente los valores antiguos y nuevos, la identificación del operador y la marca de tiempo.
Paso 4: Establecer controles de firma electrónica
Para cumplir con las expectativas de la FDA en materia de firmas electrónicas, cada usuario debe tener una identidad única. Las firmas electrónicas deben estar vinculadas a registros específicos y no deben copiarse ni reasignarse.
Paso 5: Controlar el acceso de los usuarios
Utilizar un acceso basado en roles. Los diferentes roles deben tener permisos distintos. Los administradores siempre deben tener los permisos más amplios, mientras que los operadores solo deben tener permisos parciales.
7. Ruida Packing Machinery: máquinas que cumplen con la FDA y las buenas prácticas de fabricación (GMP)
A veces, aunque los fabricantes estén dispuestos a equipar sus máquinas con un sistema de registro de auditoría, no todas las máquinas lo admiten. Ruida Packing, en colaboración con conocidas empresas farmacéuticas, entre las que se incluyen UCB y US Pharma, está sin duda comprometida con el desarrollo de equipos que cumplan con la Parte 11. Nuestras máquinas cuentan con la certificación CE, cGMP, ISO y otros certificados relacionados. Según las necesidades de los clientes, Ruida también puede proporcionar soluciones de monitorización conforme a la norma 21 CFR Parte 11 para equipos de producción y envasado.
Ruida también apoya la fabricación farmacéutica con servicios prácticos:
Materiales certificados: acero inoxidable 304 para las piezas sin contacto y acero inoxidable 316L para las piezas en contacto.
Servicios de puesta en marcha a distancia y in situ, con centros de posventa en Norteamérica y Hong Kong.
Guía completa de la máquina: manuales, instrucciones de funcionamiento, documentos de mantenimiento y vídeos de posventa.
Documentación de IQ, OQ y PQ: Asistencia opcional para documentos de validación (IQ, OQ y PQ) y documentación de cumplimiento normativo.
Para los fabricantes que tengan previsto cumplir con la norma 21 CFR Parte 11, elegir equipos con opciones preparadas para el cumplimiento normativo puede reducir los costes de modificaciones futuras y facilitar la preparación para las auditorías.
8. Conclusión
Si echamos la vista atrás a cuándo entró en vigor la norma 21 CFR Parte 11, podemos ver hasta qué punto ha marcado profundamente el sector desde 1997. Desde el 20 de agosto de ese año, esta normativa ha sido la norma que rige cómo deben gestionar los datos electrónicos las empresas reguladas por la FDA. Hoy en día, el cumplimiento de la norma 21 CFR Parte 11 es un tema central y motivo de preocupación para muchos fabricantes de productos farmacéuticos y nutricionales, por lo que es recomendable prepararse para ello. No solo en lo que respecta a la validación, sino también a la maquinaria.
9. Preguntas frecuentes
P1: ¿Es obligatoria la norma 21 CFR Parte 11?
Sí, por supuesto. Pero no se aplica a todos los sistemas de registro electrónico. Es obligatorio cuando se utilizan registros electrónicos o firmas electrónicas para cumplir los requisitos de la FDA.
P2: ¿En qué consiste la diferencia entre las BPF y la Parte 11 del 21 CFR?
Pues bien, tienen enfoques diferentes. Las BPF se centran en la calidad del producto, mientras que la norma 21 CFR Parte 11 se centra en los registros electrónicos y las firmas electrónicas.
P3: ¿Todas las máquinas farmacéuticas deben cumplir con la Parte 11?
No. Una máquina debe tener en cuenta la Parte 11 cuando crea, almacena, modifica o transmite registros electrónicos regulados. Es posible que los equipos puramente mecánicos no requieran el mismo nivel de control.
P4: ¿Qué es un registro de auditoría?
Un registro de auditoría consiste en registrar cualquier acción realizada en el sistema, indicando la acción exacta, la hora y cualquier cambio.
P5: ¿Cómo puedo saber si un equipo cumple los requisitos de la Parte 11?
Puede comprobar si admite registros de auditoría, control de acceso de usuarios, firmas electrónicas, almacenamiento seguro de datos, exportación de registros y documentos de validación. Pero, en pocas palabras, simplemente pregunte a los proveedores de la máquina antes de comprarla.
[1] Administración de Alimentos y Medicamentos de EE. UU. (FDA): Parte 11, Registros electrónicos; Firmas electrónicas — Ámbito de aplicación y uso
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
[2] Código Electrónico de Regulaciones Federales: 21 CFR Parte 11 — Registros electrónicos; Firmas electrónicas
[3] Registro Federal: Registros electrónicos; Norma definitiva sobre firmas electrónicas, publicada el 20 de marzo de 1997